Each new virus genome carries information about the history of its mutation. The mutating frequency, correlated with the sequencing results of a given virus strain, even allows the path of infection to be reconstructed. In this article, we describe the result of a bioinformatics analysis devoted to the results of sequencing SARS-CoV-2 coronavirus from around the world.
International GISAID database
The GISAID initiative was initially created to provide data on influenza viruses, but in the face of the ongoing epidemic, the database has also been adapted to the publication of information on identified SARS-CoV-2 viruses. Therefore, the results of sequencing the new coronavirus from around the world are shared in this database.
At the end of July, there were over 80,000 records in the database. The largest number of coronaviruses in the world has been sequenced this year in the UK (over 30,000), followed by the United States (over 17,000), followed by Australia and Spain, each with over 2,700 virus sequences. In Poland, the GISAID initiative in collecting the coronavirus genomes database, apart from genXone SA, was also supported by six other institutions. As a result, at the end of July, 115 coronavirus sequences from Poland were available in the database.
The coronavirus mutation on a global scale
The bioinformatics analysis shows that the SARS-CoV-2 coronavirus mutates much slower than influenza or HIV. Nevertheless, changes in its genome continue. In individual coronaviruses, we observe differences from the primary Wuhan virus in ten or more nucleotides from the full sequence of its RNA strand, and the average (median) of these differences is 8. Interestingly, in Poland alone, this average is as many as 10 nucleotides difference from the sequence of the primary virus genome from Wuhan. This may be because the virus reached us later and managed to mutate at that time.
Dr Maciej Sykulski – Scientific Coordinator of genXone explains: “The rate of mutation occurs in the current coronavirus observed in the analysis means one new nucleotide change with almost every subsequent infection. This fact gives us an advantage – it is possible to trace the chain of infections among humans only by analyzing the sequence of the viral genomes.”
Coronavirus genetic map
The compared genomes of coronaviruses allow us to create something like a family tree, on which new bifurcations arise at each moment of the virus mutation occurrence. Tracking these branches allows, first of all, to know the directions of virus flow between different regions of the world:
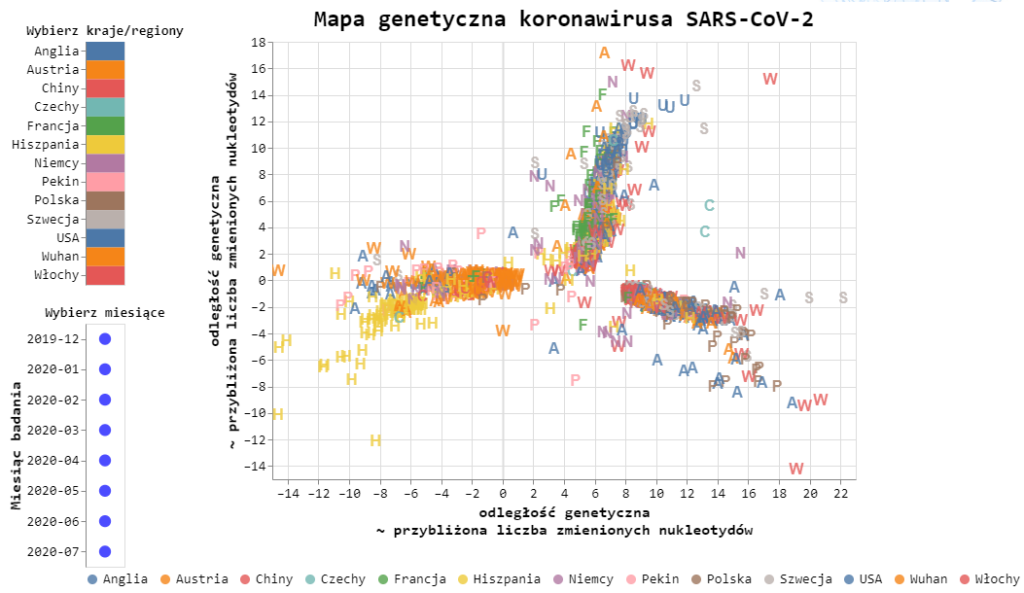
The map shows the virus branches closer to the sequence directly from Wuhan – strain B, and also found in other regions of China – strain A. Interestingly, a significant percentage of viruses in Spain originate from Chinese branches. This significantly distinguishes Spain from the European results, where strains B.1 and B.1.1 predominate. These two most abundant strains in Europe only after their European debut later found their way to other continents. The characteristic of the B.1 and B.1.1 strains is that they have mutations in the ORF1ab sequence (4715L) and the S protein (D614G) which increase the infectivity of the virus. Recent studies indicate that higher mortality is also associated with these variants.
Scientific studies from Australia, contact tracing analyses in Hong Kong, Israel, and statistical analyzes of WHO data from the start of the epidemic in Great Britain indicate that the virus is spreading heterogeneously. The phenomenon of superbugs is characteristic there, which means that 80% of infections are caused by only 10% of infected people. This way of propagating the virus is called the low dispersion factor. Our analysis of the genetic data of sequenced viruses around the world supports this hypothesis.