DNA sequencing methods produce multiple readings of different lengths. However, based solely on such results, it is not possible to obtain the comprehensive information needed about DNA molecules. The next step in the full analysis is to recreate the original DNA sequence, i.e. to know the order of the nucleotide pairs.
What is DNA assembly?
Assembling (reassembling) de novo (Latin from scratch) consists of the appropriate arrangement and connection of overlapping readings obtained from sequencing and basecalling to recreate the studied sequence from which they were created. This method is based on a reading sequence only and assumes that the reference sequence and its length are unknown in this case.
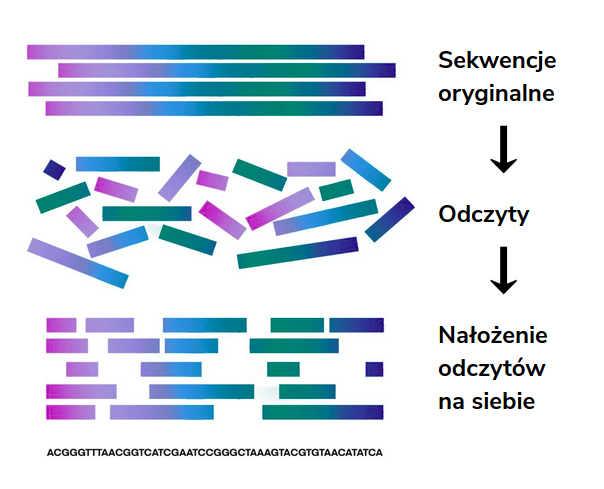
Assembling methodology and the difficulty involved
Sequencing provides a huge amount of readings that come from both strands of DNA. Therefore, it is not clear in what order the given fragments should be read, and checking all possible combinations is associated with the high complexity of the algorithm. The DNA pool under study can also come from many organisms, the readings of which must be submitted separately, which poses another challenge for assembly algorithms.
However, the main problem is the accuracy of the results obtained. When assembling a DNA sequence, the readings are significantly shorter than the target sequence. This presents additional difficulty when the result sequence has a repetition sequence since short readings obtained from sequencing often make it completely impossible to determine the number of repetitions of a given fragment.
Assembling in nanopore sequencing
Readings obtained by nanopore sequencing are even tens or hundreds of times longer than readings obtained from commonly used techniques such as Illumina. Thanks to this property, composing, especially de novo syntax, is much more efficient, requires fewer system resources, and reduces the likelihood of an assembly error. Long, single readings from nanopore sequencing are often able to cover the entire repetitive region, which allows the number of repeats to be determined with greater accuracy. In this way, long readings also help identify structural variants. Even a single reading from nanopore sequencing can contain complex structural variants that are often impossible to track and verify with short readings.
Nanopore sequencing is a development technique, and the readings obtained with its help have a different specificity compared to competing sequencing techniques. Therefore, more and more new tools dedicated to them are created. The Medaka program, which uses a machine learning algorithm based on neural networks, can be used to create consensus sequences from long readings. Many programs used to analyze results from other techniques also create sets of parameters suitable for long readings.