Metody sekwencjonowania DNA pozwalają na uzyskanie wielu odczytów o różnych długościach. Jednak bazując wyłącznie na takich wynikach nie jest możliwe uzyskanie kompleksowych, potrzebnych informacji o cząsteczkach DNA. Następnym etapem pełnej analizy jest więc odtworzenie oryginalnej sekwencji DNA, czyli poznanie kolejności występowania par nukleotydów.
Czym jest asemblacja DNA?
Asemblacja (składanie, ang. assembly) de novo (łac. od nowa) polega na odpowiednim ułożeniu i połączeniu nachodzących na siebie odczytów pozyskanych z sekwencjonowania i basecallingu w celu odtworzenia badanej sekwencji, z której powstały. Metoda ta opiera się tylko na sekwencji odczytów i zakłada, że sekwencja referencyjna i jej długość jest w tym przypadku nieznana.
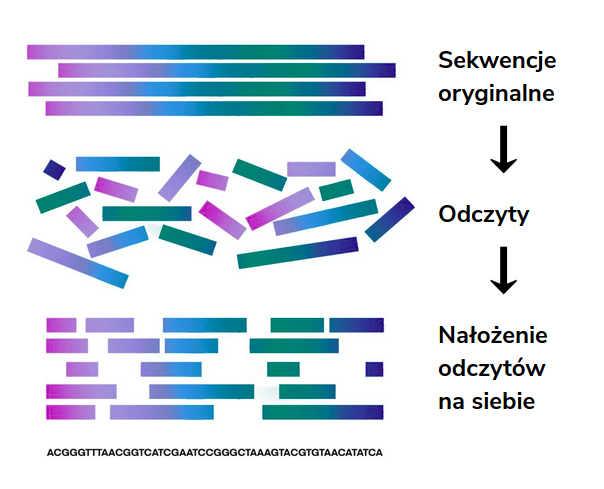
Metodyka asemblacji i występująca trudność
Sekwencjonowanie dostarcza ogromną ilość odczytów, które pochodzą z obu nici DNA. W związku z tym nie jest jednoznaczne w jakiej kolejności należy odczytywać dane fragmenty, a sprawdzenie wszystkich możliwych kombinacji wiąże się z wysoką złożonością algorytmu. Badana pula DNA może pochodzić również z wielu organizmów, których odczyty należy składać osobno, co stanowi kolejne wyzwanie dla algorytmów asemblacji.
Jednak głównym problemem jest kwestia dokładności uzyskanych wyników. Przy składaniu sekwencji DNA odczyty są znacznie krótsze od docelowej sekwencji. Sprawia to dodatkową trudność w przypadku, gdy sekwencja wynikowa posiada ciąg powtórzeniowy, ponieważ wówczas krótkie odczyty otrzymywane z sekwencjonowania często całkowicie uniemożliwiają określenie liczby powtórzeń danego fragmentu.
Asemblacja przy sekwencjonowaniu nanoporowym
Odczyty uzyskiwane za pomocą sekwencjonowania nanoporowego są nawet kilkadziesiąt lub kilkaset razy dłuższe od odczytów uzyskiwanych z powszechnie stosowanych technik jak na przykład Illumina. Dzięki tej właściwości składanie, a zwłaszcza składnie de novo, jest zdecydowanie bardziej wydajne, wymaga mniej zasobów systemowych oraz zmniejsza prawdopodobieństwo wystąpienia błędu złożenia. Długie, pojedyncze odczyty, pochodzące z sekwencjonowania nanoporowego, są często w stanie pokryć cały region repetytywny, co z większą dokładnością pozwala określić liczbę powtórzeń. Dzięki temu, długie odczyty pomagają również przy identyfikacji wariantów strukturalnych. Nawet pojedynczy odczyt z sekwencjonowania nanoporowego może obejmować złożone warianty strukturalne, które przy krótkich odczytach są często niemożliwe do namierzenia i zweryfikowania.
Sekwencjonowanie nanoporowe jest techniką rozwojową, a odczyty otrzymywane z jej pomocą posiadają inną specyfikę w porównaniu z konkurencyjnymi technikami sekwencjonowania. Dlatego też powstaje coraz więcej nowych narzędzi specjalnie im dedykowanych. Do tworzenia sekwencji konsensusowych z długich odczytów może posłużyć program Medaka, który wykorzystuje algorytm uczenia maszynowego oparty na sieciach neuronowych. Wiele programów wykorzystywanych przy analizach wyników z innych technik, tworzy też zestawy parametrów odpowiednich dla długich odczytów.